The perturbation dynamics method allows computation of differences in binding free energy between systems with complex reaction paths. We compared perturbation energy differences for three divalent cations, in the presence of water, two organic acids, or a model calcium binding, protein E. coli periplasmic glucose-galactose binding protein (GBP), to estimate of binding selectivity using the CHARMM force field (Brooks et al., 1983). The divalent cations studied were the important biological cations, Ca++ and Mg++, and the much larger Ba++, giving a cross section of ionic radii. The organic acids, a monovalent anion acetate and a trivalent anion citrate, served as controls for the parameters and methods. In addition, citrate is useful for comparison to the more rigid protein structure. These energy results are compared to experimentally measured binding.
By adjusting the parameter set to give best results, a fairly accurate perturbation method was developed. Using this method, perturbation dynamics were conducted between Ca++ and Mg++ and Ca++ and Ba++ in each of the systems listed. As support for the derived parameter set, all of the computed **G values were within 1-3 kcals of experimentally derived results. After the dynamics protocol was well set, structural analyses were conducted to make hypotheses about the source of selectivity of the EF hand site. From these analyses, it has been seen that the EF hand site provides a rigid eight fold coordinating array, in that it provided a uniform eight oxygen first shell coordination for all of the ions tested, while the optimal coordination number of each ion was found to be quite different in bulk solvent or with the acids, except for Ca++ which prefers an eight fold coordination. Other structural results support this hypothesis. From this data, we have concluded that the specificity of the site for Ca++ over larger divalent ions is a continuation of the trend observed in which higher field strength chelators are more specific for smaller ions than are lower field strength sites. For the case of exclusion of smaller radii ions, the data suggests that Mg++ is excluded from the site due to the inability of the site to provide less than an eight fold coordination array, leading to excessive electrostatic repulsion between the competing oxygens, so that all of the oxygens were unable to achieve close contact interactions with the positively charged ion. Although these conclusions can not be proved exhaustively until they are demonstrated in the laboratory, it provides a good starting point for further exploration of the mechanism of ion specificity for the site.
Selective ion binding plays a key role in diverse chemical and physiological process, including ion-selective electrodes (e.g. Eisenman, 1962; Eisenman, 1965), compleximetry (e.g. Ringbom, 1963; ), ion-selective membrane transport (e.g. Hille, 1992; Hess and Tsien, 1984; Eisenman and Horn, 1983), and intracellular signaling (e.g. Falke et al., 1994; ).
The field-strength theory of alkali metal cation binding selectivity, which was developed early by Eisenman to explain electrode selectivity (Eisenman, 1962; Eisenman, 1969) and later adopted by Hille in his ion channel selectivity filter theory (Hille, 1974), predicted binding affinity from ion and site hydration energies, which depend on ion and site field strengths. Current molecular dynamics computations using explicit water molecules model the balance between hydration and binding in better detail. This classical atomistic approach using empirical force fields fails to include the significant effects of atomic and molecular polarization. Nevertheless, it is shown to be useful here to help discern the mechanism of selectivity in a calcium binding protein.
The Eisenman theory focused on the electrostatic consequences of the size (and shape) of both the binding ion and the ligation site. It illustrated that in the asymmetry between ion-site, ion-water, and site-water interactions, low field strength sites (having less charge density at the surface) should preferentially bind larger metal cations with higher affinity whereas high field strength sites would favor smaller metal cations. Monovalent cation selectivity sequences corresponding to the eleven permutations predicted by Eisenman theory have been observed in nature. However, the theory essentially neglects steric factors such as access to or crowding in an ion binding site as well as binding-induced strain in the solute or ligand.
The cation binding properties of EF-hand sites show complexities that are thought to reflect such steric and strain factors (reviewed in Falke et al., 1994). By exploring the consequence of mutations to charge and volume of coordinating EF-hand side chains on the selectivity data for extensive divalent and trivalent cation series, they concluded that they could induce changes in the optimal radius for binding or the flexibility in coordinating larger cations (Falke et al., 1991). Additionally, some changes effectively eliminated cation binding. The reduction in size selectivity for above-optimal-radius cations with increased binding site charge is contrary to the predictions of Eisenman selectivity theory, but this and the other phenomena could be readily rationalized in terms of the steric alterations expected for the given mutations.
We have chosen to analyze the divalent cation-binding selectivity of the glucose- galactose receptor from E. coli (GBP) which is especially well suited for careful analysis of divalent binding because, as pointed out by Falke et al. (1992): a) Crystal structures of apo- and holo-structures are available; b) The binding site is an EF-hand site and therefore is relevant to a large array of other divalent binding proteins; c) The protein contains only one binding site per molecule and therefore yields an unambiguous assay of first -ion binding without the complexities of cooperativity; and d) Measurements of binding affinity for the complete series of mono-, di-, and trivalent cations have been performed with both wild type and site-mutant variants of the protein which, if the assumption of backbone homology is justified, provides ample data for comparison with computation.
Here we report the results of studies of perturbation dynamics computations for mutations from either Ca++ to Ba++ or Ca++ to Mg++ in environments consisting either of a water ball, two hydrated anions: acetate (monovalent) or citrate (trivalent), or hydrated GBP. These results illustrate that the GBP binding site is more selective for Ca (against Mg or Ba) than expected based on charged oxygen ligation alone because of strain in the protein.
The free energy perturbation method (Beveridge et al; 1989; Brooks III et al, 1988) allows determination of free energy differences between two systems with differing characteristics, allowing calculation of energy differences between reaction coordinates that have a complex connecting reaction path. Mutations between Ca++ and Ba++ or Mg++ were used as estimates of the difference in the solvation and ligation energies. The results were compared to experimental estimates of the ion binding selectivity of two common organic acids, acetate and citrate, and by the EF-hand Ca++ binding site from the E.Coli glucose-galactose receptor protein (GBP). In each case, the experimental binding selectivity was taken as DDG=-RTln(KCa/KX) where DDG is the difference in free energy of binding, R and T are the gas constant and absolute temperature, and KCa and KX are the binding constants for Ca++ and either Ba++ or Mg++ respectively (Martell and Smith). The mutations in the water ball were used to estimate the difference in solvation energy of the two ions, the left hand limb of the thermodynamic cycle.
DG1 M++ . H2O + GBP --------> M++ . GBP + H2O | | | DG3 | DG4 (1) V DG2 V Ca++ . H2O + GBP --------> Ca++ . GBP + H2O
Where DG4 - DG3 = DG2 - DG1 = DDG. This DDG reflects the chelator binding selectivity for the ion vs. Ca++. M++ represents Ba++ or Mg++.
Protein ModelThe crystal structure of GBP with Ca++ bound was taken from Brookhaven Protein Data Bank, (2GBP, Brookhaven PDB). For computational efficiency, the system was truncated by cutting all residues and implicit waters not containing atoms within 15A of the ion binding site in the crystal structure, removing most of the protein not associated directly with the Ca++ binding site. Terminal peptides resulting from these cuts were left uncharged to prevent electric field distortions and with only one H or O attached to the backbone for simplicity. The portion used contained all of the model EF-hand site including all residues containing important coordination oxygens and had 4 negative residues, including glutamic acid at positions 174 and 205, and aspartic acid at positions 134 and 138, and 5 positive residues, which were all lysine, at positions 125, 137, 172, 203 and 227. As a test of the flexibility sufficiency of this subset, we performed dynamics with the whole protein and for a test of the possible impact of acid side chain neutralization due to possible pKa shifts (Falke, 1994; Honig and Nicholls, 1995), we used the truncated binding protein with glutamine 205 replaced by its protonated analog. Vacancies in the 15A ion-centered protein sphere were filled with water molecules taken from an equilibrated sphere (Molecular Simulations Inc., (MSI) Burlington, MA). Atomic partial charges were generated from the MSI residue file AMINOH.RTF. All bond and angle parameters were taken from the MSI PARM.PRM file.
Acid and Water ModelsThe organic acids citrate and acetate were constructed using Quanta (MSI). Carboxylate and other relevant partial charges were assigned using structural analogs from the Charmm residue file, AMINOH.RTF. Thus the organic acid carboxylate groups have the same partial charge distribution as carboxylate groups in the amino acids of the model EF-hand site. Specifically, in all of the acid groups, all hydrogen atoms were assigned a charge of zero, the carbonyl carbon +.36, the alpha carbon -.16, and the acid oxygens each -.6, giving a net charge of -1. In addition, for the case of citrate, the hydroxyl oxygen was given a charge of -.65, the attached hydrogen a charge of +.4, and carbon number 5 (attached to the hydroxyl group and alpha to an acid group) a charge of +.09. For citrate and acetate, the pKa's are low enough for complete dissociation at physiological pH's (Martell and Smith). Water molecules were the modified TIPS3P water model (Jorgenson, 1983; Reiher, 1985) which includes a partial charge of -.834 on the oxygen and +.417 on the hydrogens and a pseudobond between the hydrogens.
Cation ModelThe Cations used were Ca++, the usual occupant of the EF-Hand site, as well as the much smaller sized Mg++ and the much larger Ba++ to provide a range of ion radius and selectivity. The atomic charge for each of the three ions was +2. The van der Waals Emin parameters were those given in the MSI PARM.PRM parameter file. The van der Waals radius, Rmin, for Ca++, obtained from Benoit Roux, was adjusted to produce an energy of charging equal to the hydration energy minus a small cavitation energy. With this ion radius set, the van der Waals radii for the other ions were adjusted to give appropriate hydration energy differences under free energy perturbation dynamics calculations with a 10A water ball (MSI).
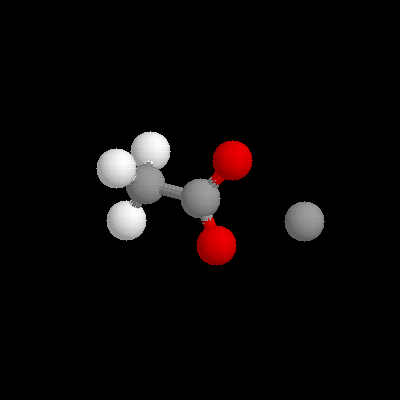
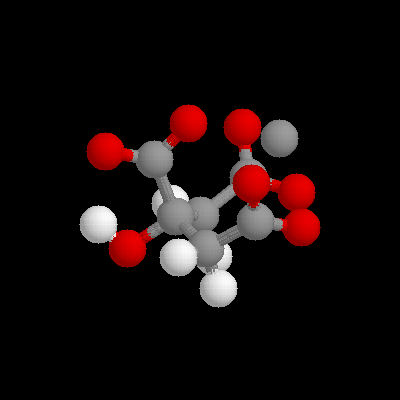
System ModelBlack and white representative pictures of the systems used can be seen in figures 1 and 2. Figure 1a shows citrate coordinating Ca++, surrounded by a 10A sphere of solvent. In figure 1b, the truncated GBP protein is depicted, with the 15A sphere of solvent extending out of the EF hand site. A close up stereo view of the EF hand site is shown in figures 2a and b, showing the eight coordinating oxygens available. The system used was an adaptation of the spherical stochastic dynamics method with the ion held to the center of mass with a weak mean force potential, an interior free Verlet dynamics sphere (8 A radius), an intermediate shell where Langevin dynamics to simulate a "heat bath" (2 A for water ball, with and without organic acid, calculations, 4 A for GBP calculations) and an outside boundary potential to keep the system spherical. For the water ball calculations, the Spherical Solvent Boundary Potential (SSBP, Beglov and Roux, 1994) was used to simulate the pressure (1 Atm) and surface de-orienting effects of an infinite bath of water around the system. The boundary potential was removed for Ca++ vs. Mg++ hydration energy calculations, where it appeared to result in excessive hysteresis between forward and backward mutation runs, perhaps due to inadequate equilibration in the 10 ps perturbation. In the GBP calculations, SSBP was not a good choice for the boundary potential, since a large portion of the protein surface was non - solvent. Therefore outside of 12A all the atoms were fixed in their positions which assured that the global protein shape retained the crystal structure and minimized computational time, while allowing local accommodations for ion size The van der Waals radii of the atoms are 2.08A for Ba++, 1.7 for Ca++ and 1.225 for Mg++. Thus the difference betwen Ba++ and Mg++ van der Waals volumes is 0.41% of the reaction sphere volume for this case. The energetics of this volume perturbation cannot be accurately estimated without a detailed study of the compressibility of this region of the protein, but it is expected to be minor compared to conformation and interaction energy effects.
Perturbation MethodPerturbation dynamics were calculated using the PERT facility in CHARMM. The thermodynamic integration (TI) perturbation method was used with 10 intermediate force fields, characterized by the proportionality constant l (Beveridge and DiCapua, 1989). Each run was started with 50 steps of Steepest Descents minimization, followed by 500 steps of Adopted Basis Newton - Raphson minimization (Brooks et al., 1983) to eliminate any maldistribution of potential energy before dynamics. This was followed in the case of the water ball sytems with 5 ps of dynamics, during which the sytem was heated to 300K. In the case of the protein system there were a total of 15 ps of preliminary dynamics, with a 100K heating for each 5 ps of equilibration. This was followed by a ten step l perturbation where the ion was mutated from Ca++ to either Mg++ or Ba++, or from Mg++ or Ba++ to Ca++. The perturbation was weighted to give smaller increments of l (and thus more steps of dynamics) in the direction of the smaller ion, due to the inverse square relationship of radius to free energy change indicated by the integrated Born equation, which gives the change in free energy with respect to a change in radius as a function of valence, z, the dielectric constants of the two phases at the interface, E2 and E1, and radius of the ion r.
dG/dr = - z^2e^2N(1/E2 - 1/E1) / (8(pi)(E-zero)r^2)
In the case of Ca++ -> Ba++ mutations, or Ba++ -> Ca++, each step of l was associated with 200 fs equilibration followed by 2.8 ps of simulation. For Ca++ -> Mg++, or vice versa, each step included 1 ps equilibration followed by 3 ps simulation to accommodate the larger radius change. The Shake algorithm was used to maintain normal lengths of covalent bonds to polar hydrogens. For each system, four runs were conducted in each direction, each with different random starting velocities, either from ion X++ to Ca++, or from Ca++ to X++, and the eight total runs for each system were then finally averaged to give final perturbation energy results. There was typically a histeresis between forward and backward runs of 0-6 kcal/mole. On the assumption that the correct value was bracketed by the two extremes, the average was used.
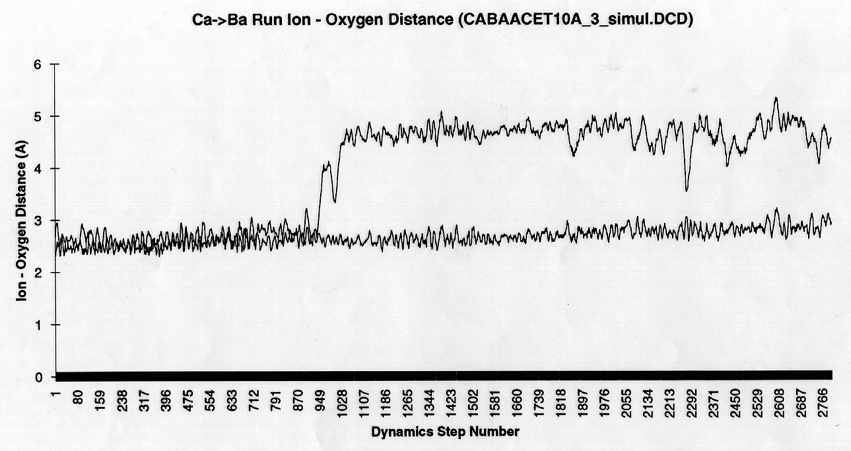
Analysis MethodsIn addition to the free energy calculations the radial density histogram of ion- oxygen distances for dynamics runs were made both during mutations of ions bound to acetate or protein and during non-perturbation dynamics with only one ion present. Graphs of ion-oxygen distance versus time for specific oxygens over the course of a mutation were also made for some of these runs. These were used to analyze the degree and kinetics of coordination over the course of a mutation.
Finally, potential energy decomposition was carried out for each of the ions in the presence of both citrate and cabp to pinpoint the source of the DDG's between the ions in the presence of these two molecules. The ensemble average of the potential energy components for ion-protein, ion-water, and water-protein interactions, and for protein and waters alone were computed from the frames of a 20 ps trajectory calculated for each of the six combinations fo one of the three ions bound to one of the two ligands.
Averaged DDGs (relative to our computed DGs of hydration) for the mutations Ca++->Mg++ and Ca++->Ba++ in each of the four systems are given in table 1b and c. DDG of binding is obtained by subtracting the mutation energy computed with the 10 A water ball from that in the bound state. Positive values reflect weaker binding by the end- product ion. As is shown in table 1b and c, all DDG values except for the aberrant Ca++- >Ba++ mutation in GBP and Ca++->Mg++ mutation in protonated GBP are within 0-3 kcals/mol of experimentally determined results (Martell and Smith; Falke et al., 1991). The computed energy of mutation in the water ball (using the SSBP boundary potential to simulate constant pressure of 1 Atm and an infinite electrostatic reaction field) was +61.4* 0.96 kcal/mol for Ca++ -> Ba++ and -77.4*2.3 kcal/mol for Ca++ -> Mg++ which can be compared to experimentally obtained differences in hydration energy of 65.7 kcal/mol and -74.7 kcal/mol respectively.
From the DDG values, the DG of Mg++ and Ba++ binding, relative to that of Ca++ binding, were determined and are plotted against ionic crystal radius (figure 3) for each of the three ligating compounds, acetate, citrate, and GBP. From Eisenman, and as observed experimentally with acetate and citrate, these graphs are expected to be monotonic increasing functions. With GPB, the computed DDG, like the experimentally measured DDG, shows an obvious deviation from this expectation for Mg++ where the DDG increases in spite of the reduced radius. More importantly, in all cases the desired 'trend' is observed, that for the mutation from Ca++ to the larger Ba++ as the chelator's field strength increases from acetate (with 1 carboxyl group), to citrate (with three carboxylates), to protein (with three carboxyls and several carbonyl oxygen's), so does the observed specificity of the chelator for Ca++ over Ba++. And, for the mutation from Ca++ to the smaller Mg++, the same trend occurs except for GPB, which does not follow Eisenman theory. The main purpose of this report is to analyse the basis for this aberration.
To explore the mechanism of the Mg++-binding anomaly, coordination analysis was conducted for each of the ions in both the acetate and the protein systems. For the acetate system, the first shell coordination number, meaning the average number of first shell interactions with oxygens increased with ionic radius as expected, 6.0 for Mg++, 7.9 for Ca++, and 9.4 for Ba++. This trend was absent in the case of the binding protein where each of the three ions was invariably coordinated by eight oxygen atoms, with the averaged values being 7.9 for Mg++, 7.9 for Ca++, and 8.0 for Ba++. These were invariably OD2 of aspartic acid 138, OE1 and OE2 of glutamic acid 205, the peptide carbonyl oxygen of glutamine 140, OE1 of glutamine 142, OD1 of asparagnine 136, and OD1 and OD2 of aspartic acid 134. In our simulations, the carboxyl group of aspartic acid 138 coordinates in a monodentate fashion, while those of aspartic acid 134 and glutamic acid 205 are bidentate. However in the crystal structure the ion is coordinated by only seven oxygen atoms, the difference being that aspartic acid 134 is mono- rather than bidentate in the crystal. Perhaps this is related to relaxation of the penatagonal bipyramid structure found in the crystal structure upon application of free dynamics or to effects of the crystal structure of carboxylate protonation.
Other qualitative results reinforced the idea of a rigid eight coordinate binding array in the protein EF-hand site. As can be seen in figure 4a, representing a graph of the two acetate carboxyl oxygen - ion distances plotted versus time (in ps) over the course of a Ca++ to Mg++ mutation, the quick and relatively unhindered transition from the bi- to monodentate state at the arrow is readily apparent. On the other hand, during the course of the same mutation in protein in figure 4b, the same transition seems to almost happen for the carboxyl groups of aspartate residue 138, but the renegade oxygen can not quite leave the solvation shell. This result reflects the rigidity of the eight fold coordination seen in the protein in solvent, as none of the coordinating oxygens are able to leave the first shell to allow better coordination of the smaller Mg++ ion. These graphs also show the tightening of the first shell structure upon mutation to Mg++, indicated by the decrease in ion oxygen distance. One might argue that time constraints may have played a role in producing the rigid coordination, but coordination changes with the organic acids were found to occur on the sub-picosecond time scale. Alternatively, there is reason for concern that our spatial constrations (fixation of all atoms more than 8 A from the binding site) may limit site flexibility. But, a control trajectory without peripheral constraints on the protein showed the same Mg++ coordination (data not shown) indicating that the observed rigid coordination was not an artefact of the model constraints. Furthermore, energy decomposition results suggest that the computed energetics are accurate as will be discussed below.
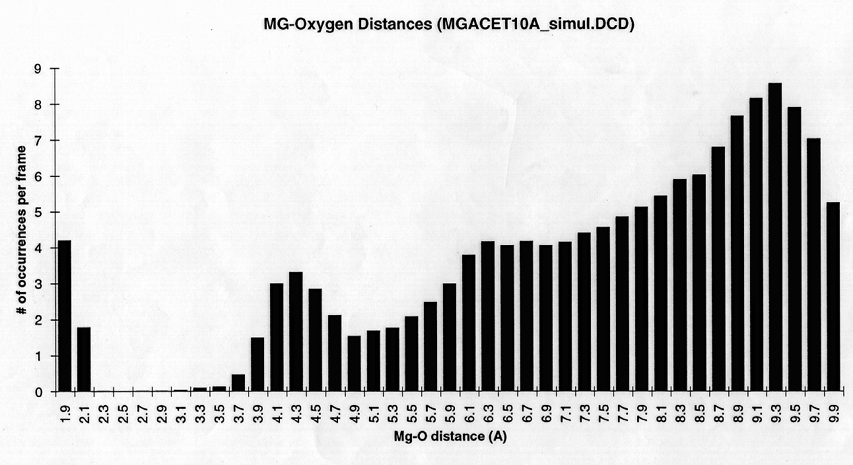
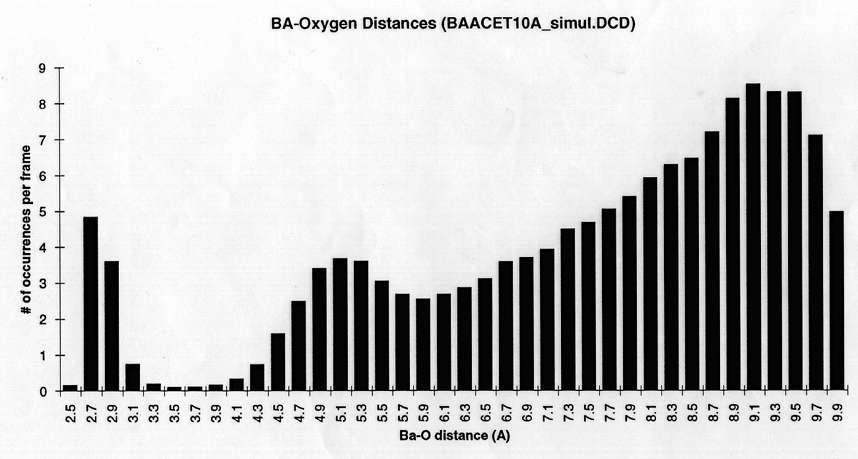
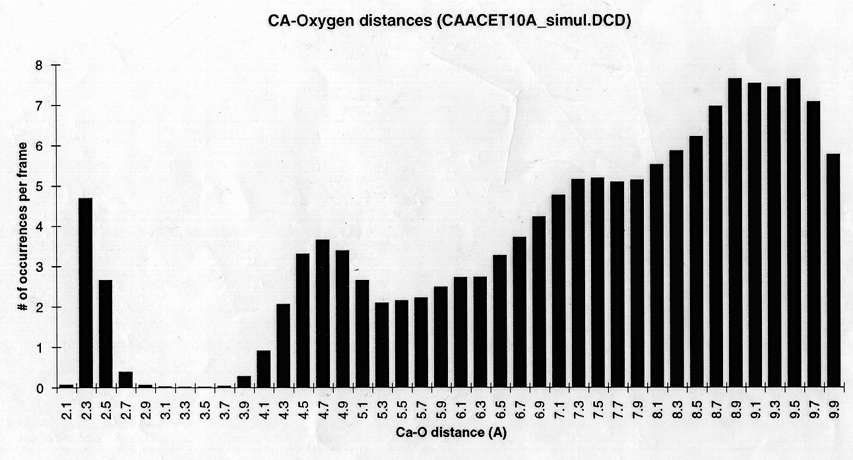
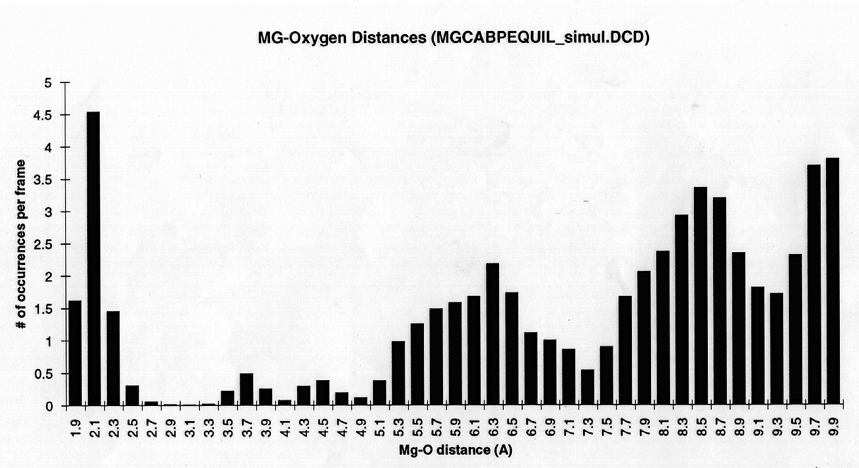
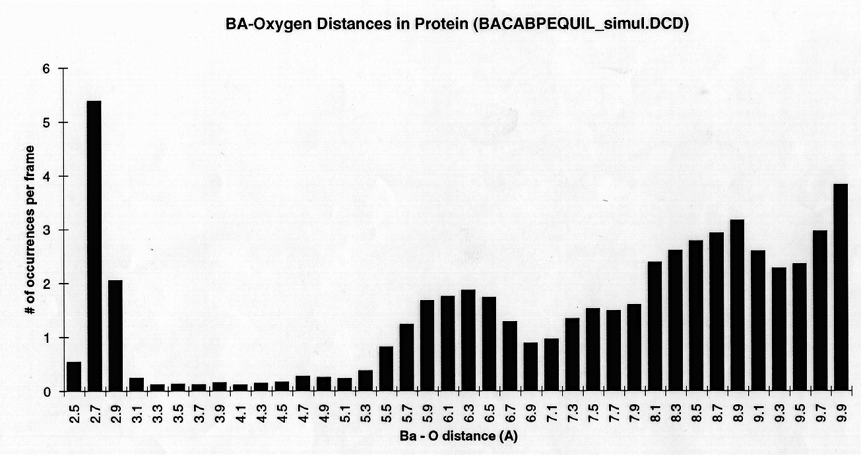
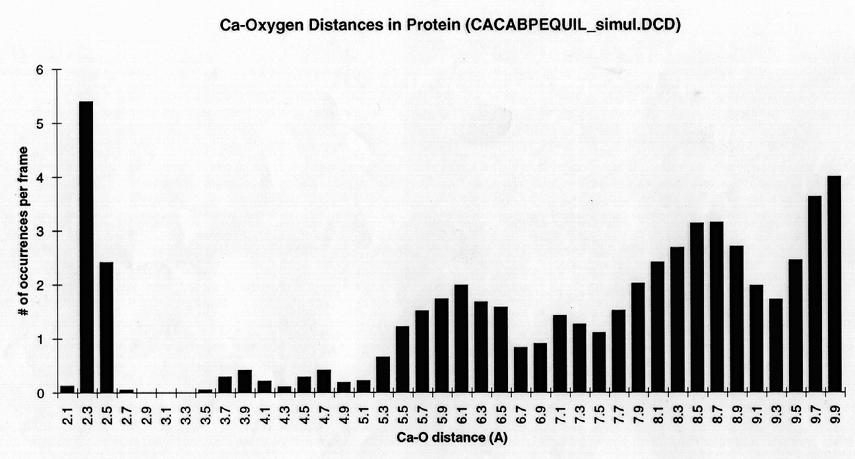
Radial density function histograms for each of the three ions coordinated either by acetate or GBP (Figure 5) highlight the difference in inner shell coordination number and mode ion-oxygen distance for the two systems. With acetate, Mg++ has a mode inner shell oxygen distance of 1.9A, Ca++ 2.3A, and Ba++ 2.7A. With GBP this value is 2.1A for Mg++, 2.3 A for Ca++ and 2.7 A for Ba++. Thus, GBP oxygens do not coordinate Mg++ optimally.
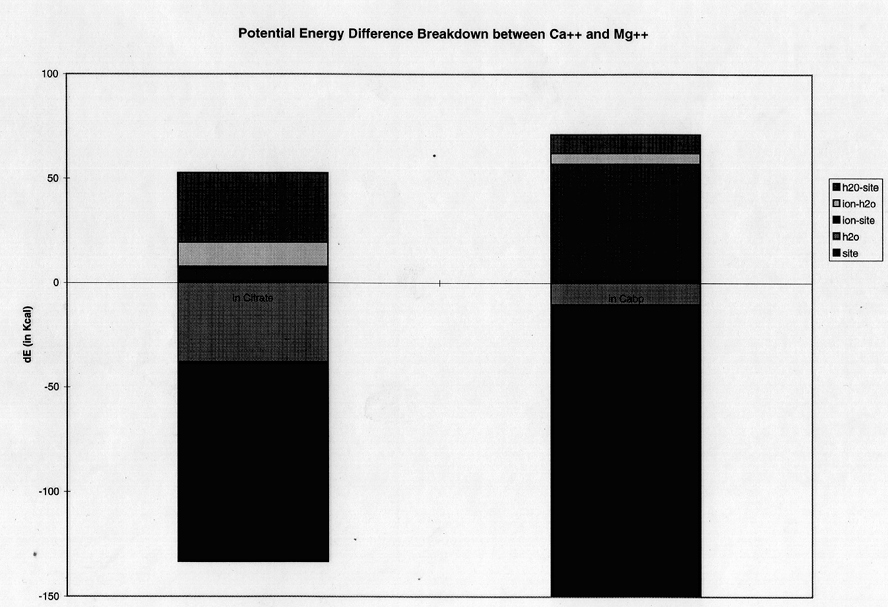
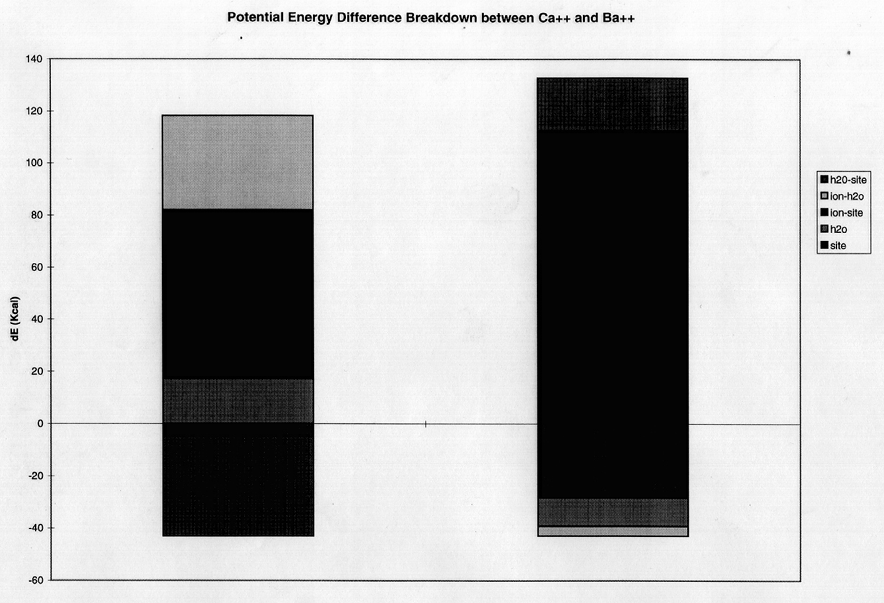
In a pairwise difference potential energy decomposition, the difference in average dynamic potential energy for Mg++ (figure 6a) or Ba++ (figure 6b) relative to Ca++ for citrate binding is compared to those for GBP-binding. These calculations were taken from the same non-perturbation dynamics trajectories used above. In each case, the largest contribution to the energy component was the electrostatic (as opposed to the van der Waals or the internal energy) term. For both ligands there is a decrease in electrostatic interaction energy between chelator and ion (ion-cit, ion-GBP) for the smaller ion. This accords with the expected specificity according to Eisenman theory of the protein for Ca++ over Ba++ (figure 6b). The trend is also seen in the Ca++ to Mg++ decomposition (figure 6a), but is more than compenstated by a parallel increase in unfavorable internal protein energy for the smaller ion. This internal protein strain stems almost exclusively from the electrostatic factor term which represents both close-range (hydrogen bond) and long- range nonbonded interactions.
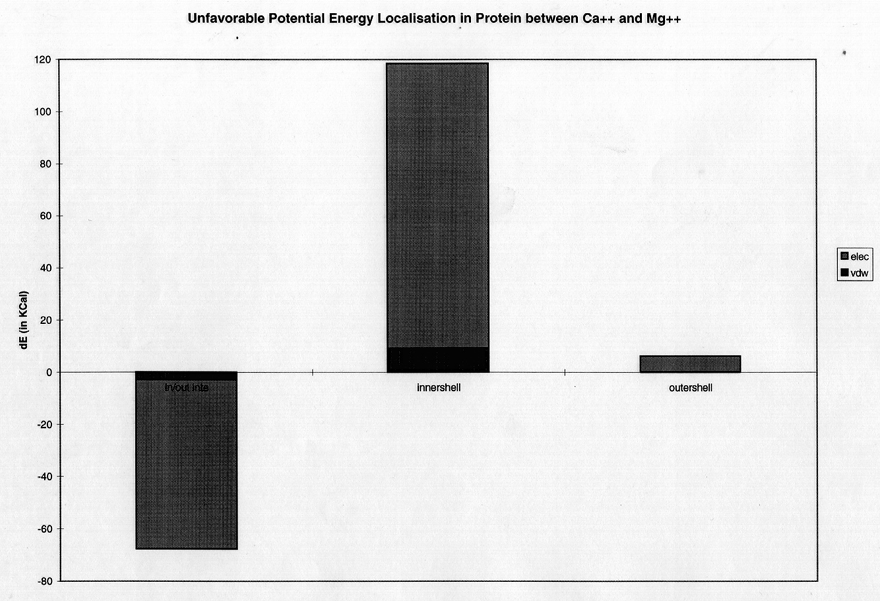
To isolate the source of this electrostatic strain in the protein, a further decomposition of the average Ca++-Mg++ difference in nonbonded interaction energy in the inner and outer coordinating shells (reflecting the eight coordinating oxygen atoms versus the rest of the protein) was performed (figure 7), again using the non-perturbation dynamics trajectories. The Mg++-induced protein strain (relative to Ca++-ligated) is subdivided into three ensemble average Ca++-Mg++ difference components: the nonbonded interaction energy of the inner shell of eight oxygens (left), that of the protein (center), and that between the inner shell and the rest of the protein (right). It is evident that the protein strain that prevents Mg++ binding (compared to Ca++) results from electrostatic overcrowding between the oxygens directly coordinating the Mg++.
The general applicability of our computational method and the CHARMM force field to real world results and findings is supported by the basic correlation of the results of our simulation to experimental results. We were able to recreate the observed trend of ionic selectivity for two acids, acetate and citrate, and for the calcium binding site of the galactose binding protein. Also, except for the extremely loose binding of Ba++ to the unprotonated protein (as compared to Ca++), all estimates of differential binding energy between ligands fell within 2-3 kcals of experimental results. These results tend to illustrate that our method provides valid energy results for these systems, leading us to suppose that structural results drawn from these simulations might also be used to make generalizations about the mechanism of ion binding specificity of galactose binding protein in solution. This hope, however, must be tempered by the fact that the underlying force fields are very simplified (e.g. neglecting atomic and molecular polarizability) and have only been minimally tested for divalent ion interactions.
The coordinating array of oxygens in the case of the acids acetate and citrate is much less rigid and well defined than for the EF hand site of the protein according to our simulations. In acetate, we observe a range of oxygen coordination numbers for the ions, ranging from ~ 6 for Mg++ to ~8 for Ca++ to ~9 for Ba++. In this case, since the carboxyl groups of acetate, as well as the coordinating water oxygens, all have considerable freedom of movement and cannot be considered greatly restricted in any way, this must indicate the optimal coordination number for each of the ions. Hence the optimal coordination number of a divalent ion is highly dependent on it's ionic radius, which limits the number of atoms able to achieve a close first shell contact with the ion. In the case of the protein, this variable coordination is lacking. Invariant of ion ligand, the protein provided a first shell coordination array of eight oxygen atoms, where none of the atoms in this array had the flexibility to leave the binding array. Moreover, the identity of the eight coordinating oxygens was consistent through the simulations or each of the ions tested, reinforcing the idea that the protein provides a rigid eightfold first shell binding array which is inflexible to any change which might accommodate larger or smaller ions.
This idea is also supported by the qualitative evidence in figures 4a and b. In the case of the acetate switch from bi to mono dentate coordination mode, the transition is quick, fluid and virtually without halt, taking place in the time span on the order of less than 200 femtoseconds. However, when the same transition seems imminent in the course of a Ca++->Mg++ mutation in binding protein, it is evident in figure 4b that the second carboxylate oxygen is simply unable to move far enough away from the ion for an extended period of time to achieve the monodentate binding mode. Each time the second oxygen moves some distance away from the ion, it quickly vibrates back close to the ion again, keeping it trapped in the bidentate state. This restriction is probably imposed by some steric constraint imposed by the protein binding site architecture. Also evident in this plot is the general constriction of the binding cavity during the course of the Ca++- >Mg++ mutation, evidenced by the general downward slope of the ion-oxygen distance graph through the course of the mutation. This reflects the oxygen atoms' attraction to the ever smaller ion bound in the cavity.
The existence of an inflexible eightfold coordinate binding site in the protein must have serious effects on the binding of the three divalent ions in the active site. In the case of smaller than optimal ions, the eight oxygens would be not all be able to all achieve the most favorable close contact distance with the ion, due to the fact that the closer the oxygens get to the ion, the more electronegative repulsion there would be among the binding atoms. In the case of the smaller ions in acetate or bulk water solution, the flexibility of the surrounding medium to reduce the number of coordinating oxygens allows them all to have optimal close contacts with the ions. In the case of larger than optimal ions, the main reason for reduced binding affinity appears to be that the protein binding site may be unable to provide the optimal number of coordinating oxygens for the larger radius ion, resulting in suboptimal binding energy between Ba++ and inner-shell oxygens. Alternatively, the protein binding site volume may be unable to accommmodate the larger volume ion, resulting in possible unfavorable van der Waal collisions. It is interesting to note that both the number of coordinating oxygens and the mode oxygen-ion distance are identical for Ca++ in protein as in acetate, indicating that the protein is ideally suited to bind Ca++.
With this background evidence in hand, we are now able to make some conjectures, based on the computational simulations, of the mechanism of specificity of Ca++-specific EF hand sites. For the case of exclusion of larger radii ions, such as Ba++, the selectivity of protein for Ca++ seems to arise as a continuation of the general trend that higher field strength chelators are generally more specific for smaller ions than are lower field strength ones. Thus, since the protein reflects a significant increase in field strength compared to citrate and acetate, it is more specific than either for Ca++ over Ba++. In addition, this effect could be exacerbated due to the fact that Ba++ is unable to be conjugated by the optimal number of oxygens in the protein, leading to 1/9 less interaction energy between the ion and inner shell oxygen atoms. This conclusion is supported in the decomposition in figure 6b, in which the main difference in the potential energy of the protein system with Ba++ present instead of Ca++ arose as a result of a 112 kcal/mol more unfavorable ion-protein interaction when Ba++ was present, almost double the equivalent value for the citrate system. This conclusion seems to contradict that of Falke (Falke et al, 1991), who proposes that the larger Ba++ ion causes a strain on the size of the cavity when introduced to the binding site, leading to van der Waals collisions and unfavorable bond stretching around the active site. Our decomposition results contradict this hypothesis, as the internal energy of protein is actually about 30 kcal/mol more favorable when Ba++ is in the active site then when Ca++ is in the active site. Also, the mode ion/inner-shell oxygen distance is the same for Ba++ in the binding protein as it is in the presence of acetate (figure 5).
The argument for the mechanism of exclusion of smaller radii ions, such as Mg++, is a bit more specialized. In this case, the relaxed EF hand binding site cavity is too big to allow all eight coordinating oxygens to achieve close contacts with the Mg++ ion, and if the atoms forming the cavity attempt to squeeze in to coordinate Mg++, electrostatic repulsion between the eight closely packed oxygen atoms results in significant unfavorable oxygen-oxygen interaction energy and limits the amount of constriction. Because of the protein structure, none of the eight oxygens can leave the first shell, so there is no way to ease this close packing of partial and valence negative charge on the oxygens. Since the binding protein is unable to provide optimal ion oxygen interaction, the smaller ion would rather remain solvated in free solvent, where it can achieve close contacts with 6 water oxygens. This hypothesis is supported by the fact that the mode ion/inner-shell oxygen distance is higher in the binding protein then in citrate (figure 5), as well as by the decomposition results (figure 6a), which show that although Mg++ in the protein has 45 kcal/mol more favorable ion-protein interaction in the site than with Ca++ present, due to the proximity oxygens are able to achieve with the small Mg++ ion, this is more than compensated for by a 57 kcal/mol more unfavorable internal protein energy with Mg++ bound, 51 kcal/mol of these being unfavorable electrostatic interactions within the protein. When the internal protein energy is decomposed further as in figure 7, it is apparent that most of this positive energy comes from unfavorable electrostatic interactions among the eight coordinating oxygen atoms, meaning that the constricted cavity is suffering from electronegative repulsion within the site. This hypothesis agrees quite well with that of Falke, who makes similar statements concerning the exclusion of smaller radii ions (Falke et al, 1991).
Although important generalizations can be made from our results, the current computational approach is not without flaw. The energies calculated contain some deviation from experimental values, partly due to statistical/sampling error, but may be also due to inadequate conformational sampling. The selectivity of all the carboxyl groups seems a bit exaggerated in most cases, possibly suggesting that the partial charges on the carboxylates conferred too high a field strength. The lack of atomic and molecular polarizability of charge may also be an important factor of unknown magnitude. But the general correlation of computational results with experimental results argues that the model does allow important conclusions to be made, and that the partial charges used must at least partially compensate for the lack of polarizability. Another issue is whether or not one of the carboxylate groups in the binding site may be partially protonated, because when one of the acidic glutamates within the active site is replaced by it's protonated analog, the excessive selectivity between Ca++ and Ba++ in the binding protein active site (table 1), is eliminated. The possibility of a protonated binding site should be explored more fully in subsequent studies.
In summary, it seems that we have a fairly reliable atomic parameter set and system model that yields correct ligation energies to within a few kcal/mole. We have been able to use the perturbation dynamics computational method to make relevant predictions and hypotheses for the mechanism of action of a naturally occurring ion binding site, and our energy decomposition and ion coordination studies provide fairly exhaustive evidence of the mechanism of specificity of this site. These results give indications of distinct hypotheses about the nature of the exclusion of both large and smaller radii ions, results that must be further proved experimentally to be considered totally relevant. Another area of future study could also involve an investigation of the protonated binding site, as preliminary results indicate this may be a more energetically accurate depiction of the site in vivo. Also, studies could be done with a more extensive ion set, including monovalent and trivalent ions, as well as additional acids, to test the expandability and generalizabilty of the system model used. In general, as computer performance improves, longer duration trajectories can be calculated and polarization effects can be introduced which should allow more accurate structure and energy simulations.
We are still working on filling in the omissions here.
Beglov, D. and B. Roux. 1994. Finite representation of an infinite bulk system: solvent boundary potential for computer simulation. J. Chem. Phys. 100:9050-9063.
Beveridge, D.L. and DiCapua, F.M. 1989. Free energy via molecular simulation: Applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 18: 431-92.
Brooks, B.B., Broccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. Copmutational Chem. 4:187-217.
Brooks III, Charles L.; Karplus, Martin; and Pettit, B.Montgomery (1988) Proteins: A Theoretical Perspective of Dynamics, Structure and Thermodynamics. John Wiley and Sons. New York.
Eisenman, G. 1962. Cation selective glass electrodes and their mode of operation. Biophys. J. 2 (Part 2): 259-323.
Eisenman, G., and R. Horn. 1983. Ionic selectivity revisited: the role of kinetic and equilibrium processes in ion permeation through channels. J. Mol. Biol. 767:197-225.
Falke, J. J., Snyder, E. E., Thatcher, K. C., and Voertler, C.S. 1991. Quantitating and engineering the ion specificity of an EF-Hand-like Ca2+ binding site. Biochem. 30: 8690- 8697.
Falke, J. J., Drake, S. K., Hazard, A. L., and Peersen, O. B. 1994. Molecular tuning of ion binding to calcium signaling proteins. Quart. Rev. of Biophys. 27: 219-290.
Hille, B. 1974.
Honig, B. and Nicholls, A. 1995. Classical electrostatics in biology and chemistry. Science 268: 1144-1149.
Jorgensen
Reiher (see Feller et al)
Martell and Smith Critical Stability Constants
figure 1a) Zoomed out view of citrate system, surrounded by 10A water ball. b) Zoomed out view of protein system, with 15A solvent sphere extending out of the active site.
table 1 Perturbation Results a) The actual perturbation energies for mutations from Ca++- >Mg++ and Ca++->Ba++ which reflect the free enrgy change as one goes from each system with Ca++ present to the other ion. These are the average of eight runs, four in each direction, as described in text. b) Table of dG of binding for the Ca++->Ba++ mutation for each fo the four systems, derived from both experimental and computational results for comparison. c) same as above, but for the Ca++->Mg++ mutation.
figure 2) Stereo view of ef-hand site used in calculations showing coordinating oxygens.
figure 3) Graph of dG of binding versus effective ionic radius. Values for dG of binding are shown as a function of ionic radius for the three chelators, with data plotted for each of the three ions. The dG of binding for Ca+ is defined as an arbitrary zero point to provide a reference frame for the other ions.
figure 4 a) Graph of ion-oxygen distance versus reaction coordinate for Ca->Ba mutation in acetate, showing the proximity of the two carboxyl oxygens to the ion. b) The same graph, but for a Ca->Mg mutation and showing a carboxyl group in the protein.
figure 5 (a-c) Histograms of ion-Oxygen distance for all oxygens for each ion equilibrating in the presence of acteate and solvent over the course of a 20ps simulation. (d-f) Same as above, but for the protein system.
figure 6 a) Component breakdown into internal protein and water energy differences and ion-water, ion-protein and water-protein interaction energy differences for a mutation from Ca++ to Mg++ compared in the presence of protein and citrate. b) Same as above, but for Ca++ to Ba++ mutation.
Figure 7) Localisation into inner shell energy, outer shell energy and inner shell/outer shell interaction energy components of the unfavorable protein energy difference of 56 kcal between Ca++ and Mg++.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}